

IMPORTANT SAFETY INFORMATION
Hemorrhage - CYRAMZA increased the risk of
hemorrhage and gastrointestinal hemorrhage, including Grade ≥3
hemorrhagic events. In 2137 patients with various cancers
treated with CYRAMZA, the incidence of all Grade hemorrhage
ranged from 13-55%. Grade 3-5 hemorrhage incidence ranged from
2-5%.
- Patients with gastric cancer receiving nonsteroidal anti-inflammatory drugs (NSAIDs) were excluded from enrollment in REGARD and RAINBOW; therefore, the risk of gastric hemorrhage in CYRAMZA-treated patients with gastric tumors receiving NSAIDs is unknown.
- Patients with NSCLC receiving therapeutic anticoagulation or with evidence of major airway invasion by cancer were excluded from REVEL. In addition, patients with NSCLC with a recent history of gross hemoptysis, those receiving chronic therapy with NSAIDs or other anti-platelet therapy other than once daily aspirin or with radiographic evidence of major blood vessel invasion or intratumor cavitation were excluded from REVEL and RELAY; therefore the risk of pulmonary hemorrhage in these groups of patients is unknown.
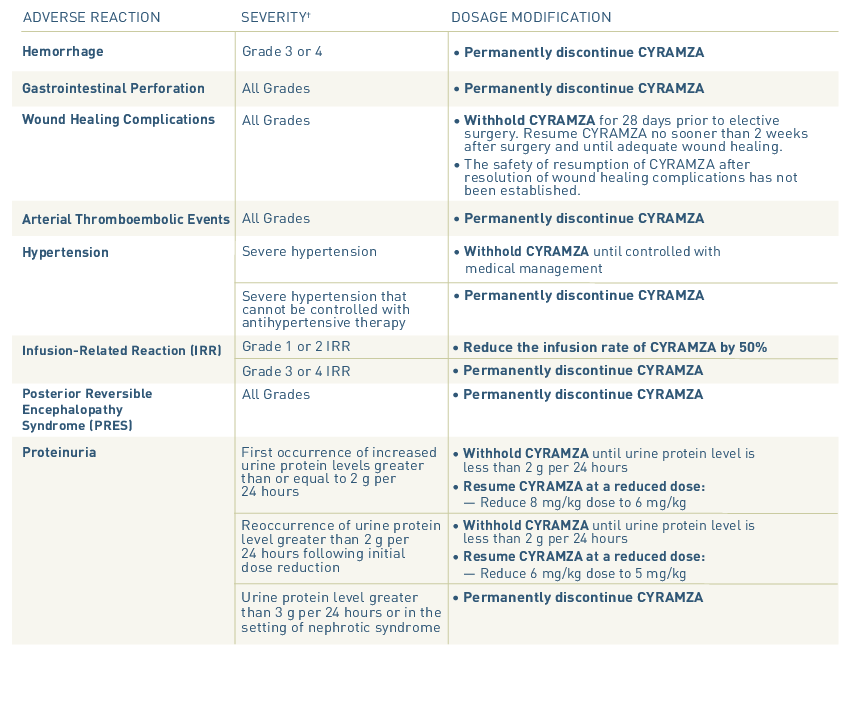
- Permanently discontinue CYRAMZA in patients who experience severe (Grade 3 or 4) bleeding.
Gastrointestinal Perforations - CYRAMZA can increase the risk of gastrointestinal perforation, a potentially fatal event. In 2137 patients with various cancers treated with CYRAMZA, the incidence of all Grade and Grade 3-5 gastrointestinal perforations ranged from <1-2%.
- Permanently discontinue CYRAMZA in patients who experience a gastrointestinal perforation.
Impaired Wound Healing - CYRAMZA has the potential to adversely affect wound healing. CYRAMZA has not been studied in patients with serious or non-healing wounds.
- Withhold CYRAMZA for 28 days prior to elective surgery. Do not administer CYRAMZA for at least 2 weeks following a major surgical procedure and until adequate wound healing. The safety of resumption of CYRAMZA after resolution of wound healing complications has not been established.
Arterial Thromboembolic Events (ATEs) - Serious, sometimes fatal, ATEs, including myocardial infarction, cardiac arrest, cerebrovascular accident, and cerebral ischemia, occurred across clinical trials. In 2137 patients with various cancers treated with CYRAMZA, the incidence of all Grade ATE was 1-3%. Grade 3-5 ATE incidence was <1-2%.
- Permanently discontinue CYRAMZA in patients who experience an ATE.
Hypertension - An increased incidence of severe hypertension occurred in patients receiving CYRAMZA. Across five clinical studies, excluding RELAY, in 1916 patients with various cancers treated with CYRAMZA, the incidence of all Grade hypertension ranged from 11-26%. Grade 3-5 hypertension incidence ranged from 6-15%. In 221 patients with NSCLC receiving CYRAMZA in combination with erlotinib in the RELAY study, the incidence of new or worsening hypertension was higher (45%), as was the incidence of Grade 3-5 hypertension (24%). Of the patients experiencing new or worsening hypertension in RELAY (N=100 CYRAMZA and erlotinib; N=27 placebo and erlotinib), 13% of those treated with CYRAMZA and erlotinib required initiation of 3 or more antihypertensive medications compared to 4% of patients treated with placebo and erlotinib.
- Control hypertension prior to initiating treatment with CYRAMZA. Monitor blood pressure every two weeks or more frequently as indicated during treatment. Withhold CYRAMZA for severe hypertension until medically controlled. Permanently discontinue CYRAMZA for medically significant hypertension that cannot be controlled with antihypertensive therapy or in patients with hypertensive crisis or hypertensive encephalopathy.
Infusion-Related Reactions (IRR) - including severe and life-threatening IRR, occurred in CYRAMZA clinical trials. Symptoms of IRR included rigors/tremors, back pain/spasms, chest pain and/or tightness, chills, flushing, dyspnea, wheezing, hypoxia, and paresthesia. In severe cases, symptoms included bronchospasm, supraventricular tachycardia, and hypotension. In 2137 patients with various cancers treated with CYRAMZA in which premedication was recommended or required, the incidence of all Grade IRR ranged from <1- 9%. Grade 3-5 IRR incidence was <1%.
- Premedicate prior to each CYRAMZA infusion. Monitor patients during the infusion for signs and symptoms of IRR in a setting with available resuscitation equipment. Reduce the infusion rate by 50% for Grade 1-2 IRR. Permanently discontinue CYRAMZA for Grade 3- 4 IRR.
Worsening of Pre-existing Hepatic Impairment - Clinical deterioration, manifested by new onset or worsening encephalopathy, ascites, or hepatorenal syndrome, was reported in patients with Child-Pugh B or C cirrhosis who received single agent CYRAMZA. Use CYRAMZA in patients with Child-Pugh B or C cirrhosis only if the potential benefits of treatment are judged to outweigh the risks of clinical deterioration.
- Based on safety data from REACH-2, in patients with Child-Pugh A liver cirrhosis, the pooled incidence of hepatic encephalopathy and hepatorenal syndrome was higher for patients who received CYRAMZA (6%) compared to patients who received placebo (0%).
Posterior Reversible Encephalopathy Syndrome (PRES) - (also known as Reversible Posterior Leukoencephalopathy Syndrome [RPLS]) has been reported in <0.1% of 2137 patients with various cancers treated with CYRAMZA. Symptoms of PRES include seizure, headache, nausea/vomiting, blindness, or altered consciousness, with or without associated hypertension.
- Permanently discontinue CYRAMZA in patients who develop PRES. Symptoms may resolve or improve within days, although some patients with PRES can experience ongoing neurologic sequelae or death.
Proteinuria Including Nephrotic Syndrome - In 2137 patients with various cancers treated with CYRAMZA, the incidence of all Grade proteinuria ranged from 3-34%. Grade ≥3 proteinuria (including 4 patients with nephrotic syndrome) incidence ranged from <1-3%.
- Monitor for proteinuria. Withhold CYRAMZA for urine protein levels that are 2 or more grams over 24 hours. Reinitiate CYRAMZA at a reduced dose once the urine protein level returns to less than 2 grams over 24 hours. Permanently discontinue CYRAMZA for urine protein levels greater than 3 grams over 24 hours or in the setting of nephrotic syndrome.
Thyroid Dysfunction - In 2137 patients with various cancers treated with CYRAMZA, the incidence of Grade 1-2 hypothyroidism ranged from <1-3%; there were no reports of Grade 3-5 hypothyroidism. Monitor thyroid function during treatment with CYRAMZA.
Embryo-Fetal Toxicity - CYRAMZA can cause fetal harm when administered to pregnant women. Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with CYRAMZA and for 3 months after the last dose.
Lactation - Because of the potential risk for serious adverse reactions in breastfed children from ramucirumab, advise women not to breastfeed during treatment with CYRAMZA and for 2 months after the last dose.
Adverse Reactions
REGARD:
- The most common adverse reactions (all Grades) observed in single agent CYRAMZA-treated gastric cancer patients at a rate of ≥5% and ≥2% higher than placebo were hypertension (16% vs 8%), diarrhea (14% vs 9%), headache (9% vs 3%), and hyponatremia (6% vs 2%).
- The most common serious adverse reactions with CYRAMZA were anemia (3.8%) and intestinal obstruction (2.1%). Red blood cell transfusions were given to 11% of CYRAMZA-treated patients vs 8.7% of patients who received placebo.
- Clinically relevant adverse reactions reported in ≥1% and <5% of CYRAMZA-treated patients in REGARD were: neutropenia (4.7%), epistaxis (4.7%), rash (4.2%), intestinal obstruction (2.1%), and arterial thromboembolic events (1.7%).
- Across clinical trials of CYRAMZA administered as a single agent, clinically relevant adverse reactions (including Grade ≥3) reported in CYRAMZA-treated patients included proteinuria, gastrointestinal perforation, and IRR. In REGARD, according to laboratory assessment, 8% of CYRAMZA-treated patients developed proteinuria vs 3% of placebo-treated patients. Two patients discontinued CYRAMZA due to proteinuria. The rate of gastrointestinal perforation in REGARD was 0.8% and the rate of IRR was 0.4%.
RAINBOW:
- The most common adverse reactions (all grades) observed in patients treated with CYRAMZA with paclitaxel at a rate of ≥5% and ≥2% higher than placebo with paclitaxel were fatigue/asthenia (57% vs 44%), neutropenia (54% vs 31%), diarrhea (32% vs 23%), epistaxis (31% vs 7%), hypertension (25% vs 6%), peripheral edema (25% vs 14%), stomatitis (20% vs 7%), proteinuria (17% vs 6%), thrombocytopenia (13% vs 6%), hypoalbuminemia (11% vs 5%), and gastrointestinal hemorrhage events (10% vs 6%).
- The most common serious adverse reactions with CYRAMZA with paclitaxel were neutropenia (3.7%) and febrile neutropenia (2.4%); 19% of patients who received CYRAMZA with paclitaxel received granulocyte colony-stimulating factors.
- Adverse reactions resulting in discontinuation of any component of the CYRAMZA with paclitaxel combination in ≥2% of patients in RAINBOW were neutropenia (4%) and thrombocytopenia (3%).
- Clinically relevant adverse reactions reported in ≥1% and <5% of patients receiving CYRAMZA with paclitaxel were sepsis (3.1%), including 5 fatal events, and gastrointestinal perforations (1.2%), including 1 fatal event.
REVEL:
- The most common adverse reactions (all Grades) observed in patients treated with CYRAMZA with docetaxel at a rate of ≥5% and ≥2% higher than placebo with docetaxel were neutropenia (55% vs 46%), fatigue/asthenia (55% vs 50%), stomatitis/mucosal inflammation (37% vs 19%), epistaxis (19% vs 7%), febrile neutropenia (16% vs 10%), peripheral edema (16% vs 9%), thrombocytopenia (13% vs 5%), lacrimation increased (13% vs 5%), and hypertension (11% vs 5%).
- The most common serious adverse reactions with CYRAMZA with docetaxel were febrile neutropenia (14%), pneumonia (6%), and neutropenia (5%). The use of granulocyte colony-stimulating factors was 42% in CYRAMZA with docetaxel- treated patients versus 37% in patients who received placebo with docetaxel.
- Treatment discontinuation due to adverse reactions occurred more frequently in CYRAMZA with docetaxel-treated patients (9%) than in placebo with docetaxel-treated patients (5%). The most common adverse reactions leading to treatment discontinuation of CYRAMZA were IRR (0.5%) and epistaxis (0.3%).
- For patients with non-squamous histology, the overall incidence of pulmonary hemorrhage was 7% and the incidence of Grade ≥3 pulmonary hemorrhage was 1% for CYRAMZA with docetaxel compared to 6% overall incidence and 1% for Grade ≥3 pulmonary hemorrhage for placebo with docetaxel. For patients with squamous histology, the overall incidence of pulmonary hemorrhage was 10% and the incidence of Grade ≥3 pulmonary hemorrhage was 2% for CYRAMZA with docetaxel compared to 12% overall incidence and 2% for Grade ≥3 pulmonary hemorrhage for placebo with docetaxel.
- Clinically relevant adverse reactions reported in ≥1% and <5% of CYRAMZA with docetaxel-treated patients in REVEL were hyponatremia (4.8%) and proteinuria (3.3%).
RELAY:
- The most common adverse reactions (all Grades) observed in patients treated with CYRAMZA with erlotinib at a rate of ≥5% and ≥2% higher than placebo with erlotinib were infections (81% vs 76%), diarrhea (70% vs 71%), hypertension (45% vs 12%), stomatitis (42% vs 36%), alopecia (34% vs 20%), epistaxis (34% vs 12%), proteinuria (34% vs 8%), peripheral edema (23% vs 4%), headache (15% vs 7%), gastrointestinal hemorrhage (10% vs 3%), gingival bleeding (9% vs 1%), and pulmonary hemorrhage (7% vs 2%).
- The most common serious adverse reactions with CYRAMZA with erlotinib were pneumonia (3.2%), cellulitis (1.8%), and pneumothorax (1.8%). Red blood cell transfusions were given to 3.2% of CYRAMZA-treated patients versus 0 patients who received placebo.
- Treatment discontinuation of all study drugs due to adverse reactions occurred in 13% of CYRAMZA with erlotinib-treated patients, with increased alanine aminotransferase (1.4%) and paronychia (1.4%) being the most common. The most common adverse reactions leading to treatment discontinuation of CYRAMZA were proteinuria (8.6%) and hyperbilirubinemia (6%).
- Of the 221 patients who received CYRAMZA with erlotinib, 119 (54%) were 65 and over, while 29 (13%) were 75 and over. Adverse reactions occurring at a 10% or higher incidence in patients receiving CYRAMZA with erlotinib and with a 10% or greater difference between patients aged 65 or older compared to patients aged less than 65 years were: diarrhea (75% versus 65%), hypertension (50% versus 40%), increased ALT (49% versus 35%), increased AST (49% versus 33%), stomatitis (46% versus 36%), decreased appetite (32% versus 19%), dysgeusia (23% versus 12%), and weight loss (19% versus 6%).
RAISE:
- The most common adverse reactions (all Grades) observed in patients treated with CYRAMZA with FOLFIRI at a rate of ≥5% and ≥2% higher than placebo with FOLFIRI were diarrhea (60% vs 51%), neutropenia (59% vs 46%), decreased appetite (37% vs 27%), epistaxis (33% vs 15%), stomatitis (31% vs 21%), thrombocytopenia (28% vs 14%), hypertension (26% vs 9%), peripheral edema (20% vs 9%), proteinuria (17% vs 5%), palmar-plantar erythrodysesthesia syndrome (13% vs 5%), gastrointestinal hemorrhage events (12% vs 7%), and hypoalbuminemia (6% vs 2%). Twenty percent of patients treated with CYRAMZA with FOLFIRI received granulocyte colony- stimulating factors.
- The most common serious adverse reactions with CYRAMZA with FOLFIRI were diarrhea (3.6%), intestinal obstruction (3.0%), and febrile neutropenia (2.8%).
- Treatment discontinuation of any study drug due to adverse reactions occurred more frequently in CYRAMZA with FOLFIRI-treated patients (29%) than in placebo with FOLFIRI-treated patients (13%). The most common adverse reactions leading to discontinuation of any component of CYRAMZA with FOLFIRI as compared to placebo with FOLFIRI were neutropenia (12.5% vs 5.3%) and thrombocytopenia (4.2% vs 0.8%). The most common adverse reactions leading to treatment discontinuation of CYRAMZA were proteinuria (1.5%), and gastrointestinal perforation (1.7%).
- Clinically relevant adverse reaction reported in ≥1% and <5% of patients receiving CYRAMZA with FOLFIRI was gastrointestinal perforation (1.7%), including 4 fatal events.
- Thyroid-stimulating hormone (TSH) levels were evaluated in 224 patients (115 CYRAMZA with FOLFIRI-treated patients and 109 placebo with FOLFIRI-treated patients) with normal baseline TSH levels. Increased TSH levels were observed in 53 (46%) patients treated with CYRAMZA with FOLFIRI compared with 4 (4%) patients treated with placebo with FOLFIRI.
REACH-2:
- The most common adverse reactions (all Grades) observed in single agent CYRAMZA-treated HCC patients at a rate of ≥10% and ≥2% higher than placebo were fatigue (36% vs 20%), peripheral edema (25% vs 14%), hypertension (25% vs 13%), abdominal pain (25% vs 16%), decreased appetite (23% vs 20%), proteinuria (20% vs 4%), nausea (19% vs 12%), ascites (18% vs 7%), headache (14% vs 5%), epistaxis (14% vs 3%), insomnia (11% vs 6%), pyrexia (10% vs 3%), vomiting (10% vs 7%), and back pain (10% vs 7%).
- The most common serious adverse reactions with CYRAMZA were ascites (3%) and pneumonia (3%).
- Treatment discontinuations due to adverse reactions occurred in 18% of CYRAMZA-treated patients, with proteinuria being the most frequent (2%).
- Clinically relevant adverse reactions reported in ≥1% and <10% of CYRAMZA-treated patients in REACH-2 were IRR (9%), hepatic encephalopathy (5%) including 1 fatal event, and hepatorenal syndrome (2%) including 1 fatal event.
Please click for full Prescribing Information for CYRAMZA.
RB-P HCP ISI 14SEP2022