REVEL Safety Profile
Adverse Reactions Occurring in ≥5% of Patients with a ≥2% Difference Between Arms in REVEL

*Neutropenia, thrombocytopenia, and hypertension are consolidated terms.
The following safety results are from the REVEL trial. Adverse reactions occurring in 5 percent or more of patients and with a 2 percent or more difference between arms.
The all grades adverse reactions reported for the CYRAMZA plus docetaxel arm (n equals 627) were neutropenia (55 percent), febrile neutropenia (16 percent), thrombocytopenia (13 percent), fatigue/asthenia (55 percent), peripheral edema (16 percent), stomatitis/mucosal inflammation (37 percent), epistaxis (19 percent), lacrimation increased (13 percent) and hypertension (11 percent).
The Grades 3 to 4 adverse reactions reported for the CYRAMZA plus docetaxel arm were neutropenia (49 percent), febrile neutropenia (16 percent), thrombocytopenia (3 percent), fatigue/asthenia (14 percent), peripheral edema (0 percent), stomatitis/mucosal inflammation (7 percent), epistaxis (less than 1 percent), lacrimation increased (less than 1 percent) and hypertension (6 percent).
The all grades adverse reactions reported for the placebo plus docetaxel arm were neutropenia (46 percent), febrile neutropenia (10 percent), thrombocytopenia (5 percent), fatigue/asthenia (50 percent), peripheral edema (9 percent), stomatitis/mucosal inflammation (19 percent), epistaxis (7 percent), lacrimation increased (5 percent) and hypertension (5 percent).
The Grades 3 to 4 adverse reactions reported for the placebo plus docetaxel arm (n equals 618) were neutropenia (40 percent), febrile neutropenia (10 percent), thrombocytopenia (less than 1 percent), fatigue/asthenia (11 percent), peripheral edema (less than 1 percent), stomatitis/mucosal inflammation (2 percent), epistaxis (less than 1 percent), lacrimation increased (0 percent) and hypertension (2 percent).
Neutropenia, thrombocytopenia, and hypertension are consolidated terms.
-
31% of patients (195 out of 627) in the REVEL trial received CYRAMZA for at least 6 months1
- Median duration of exposure was 3.5 months (median of 4.5 doses)
-
Discontinuation of CYRAMZA/placebo due to ≥1 TEAE was 1.4% (n=9) in the CYRAMZA plus docetaxel arm vs 1% (n=6) in the placebo plus docetaxel arm. Discontinuation of docetaxel due to ≥1 TEAE was 7.8% (n=49) vs 4.2% (n=26)2
- Treatment discontinuation due to adverse reactions occurred more frequently in CYRAMZA plus docetaxel-treated patients (9%) than in placebo plus docetaxel-treated patients (5%)
- The median relative dose intensity was 98.0% for CYRAMZA and 98.7% for placebo. The median relative dose intensity of docetaxel was 93.5% for the CYRAMZA plus docetaxel arm and 96.5% for placebo plus docetaxel arm3
- 51% (n=320/628) of patients received ≥1 subsequent therapy post-discontinuation with CYRAMZA plus docetaxel vs 55% (n=343/625) of patients for placebo plus docetaxel4
- The most common serious adverse reactions in patients who received CYRAMZA with docetaxel were febrile neutropenia (14%), pneumonia (6%), and neutropenia (5%). The use of granulocyte colony-stimulating factors was 42% in CYRAMZA with docetaxel-treated patients versus 37% in patients who received placebo with docetaxel.
- The most common adverse reactions leading to treatment discontinuation of CYRAMZA were infusion-related reaction (0.5%) and epistaxis (0.3%). For patients with non-squamous histology, the overall incidence of pulmonary hemorrhage was 7% and the incidence of Grade ≥3 pulmonary hemorrhage was 1% for CYRAMZA with docetaxel compared to 6% overall incidence and 1% for Grade ≥3 pulmonary hemorrhage for placebo with docetaxel. For patients with squamous histology, the overall incidence of pulmonary hemorrhage was 10% and the incidence of Grade ≥3 pulmonary hemorrhage was 2% for CYRAMZA with docetaxel compared to 12% overall incidence and 2% for Grade ≥3 pulmonary hemorrhage for placebo with docetaxel. Treatment discontinuation due to adverse reactions occurred more frequently in CYRAMZA with docetaxel-treated patients (9%) than in placebo with docetaxel-treated patients (5%).
- The most common adverse reactions (all grades) observed in CYRAMZA with docetaxel-treated patients at a rate of ≥30% and ≥2% higher than placebo with docetaxel were neutropenia, fatigue/asthenia, and stomatitis/mucosal inflammation.
- Clinically relevant adverse drug reactions reported in ≥1% and <5% of CYRAMZA with docetaxel-treated patients in REVEL were hyponatremia (4.8%) and proteinuria (3.3%).
The rate of adverse events* observed in the rapidly progressing disease† subpopulations was consistent with that observed in the overall safety population‡5,6
For patients in the overall safety population, discontinuation rates due to TEAEs were 9% (n=58) in the CYRAMZA plus docetaxel arm vs 5% (n=32) in the placebo plus docetaxel arm.6
For patients with rapidly progressing disease,† discontinuation rates due to TEAEs were 5% (n=5) in the CYRAMZA + docetaxel arm vs 3% (n=3) in the placebo + docetaxel arm.5
SAFETY OVERVIEW FOR THE OVERALL SAFETY POPULATION6
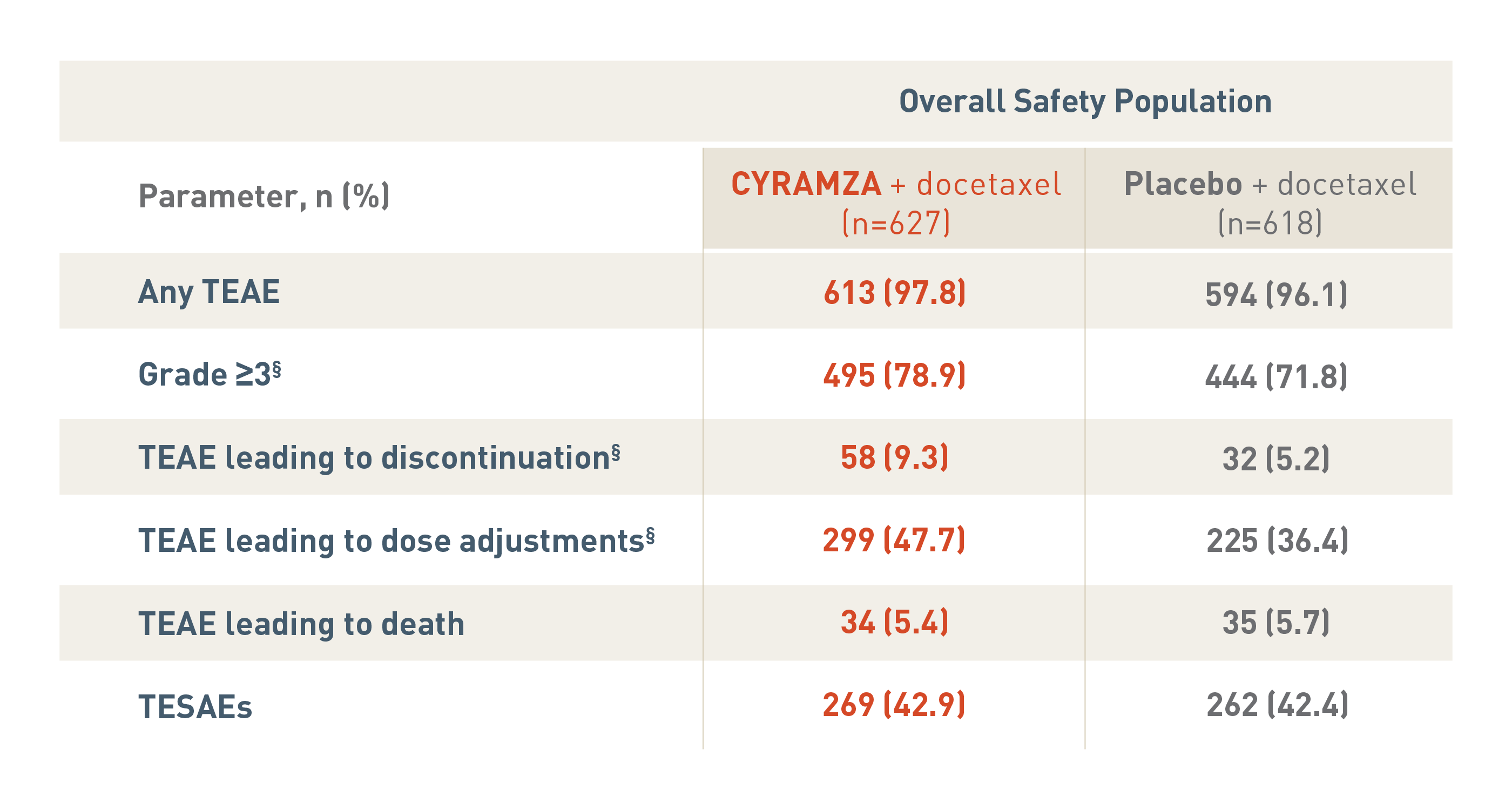
The following safety overview is for the overall safety population for the REVEL trial.
With CYRAMZA plus docetaxel (n equals 627), 613 patients (97.8 percent) experienced treatment-emergent adverse events (TEAEs), 495 patients (78.9 percent) experienced TEAEs that were Grade 3 or more with a, 58 patients (9.3 percent) experienced TEAEs leading to treatment discontinuation, 299 patients (47.7 percent) experienced TEAEs leading to dose adjustments, 34 patients (5.4 percent) experienced TEAEs leading to death, and 269 patients (42.9 percent) experienced treatment-emergent serious adverse events (TESAEs).
With placebo plus docetaxel (n equals 618), 594 patients (96.1 percent) experienced TEAEs, 444 patients (71.8 percent) experienced TEAEs that were Grade 3 or more, 32 patients (5.2 percent) experienced TEAEs leading to treatment discontinuation, 225 patients (36.4 percent) experienced TEAEs leading to dose adjustments, 35 patients (5.7 percent) experienced TEAEs leading to death, and 262 patients (42.4 percent) experienced TESAEs.
The overall incidence of TEAEs that were Grade 3 or more, TEAEs leading to treatment discontinuation and TEAEs leading to dose adjustments were significantly different between treatment arms each with a P value of less than 0.05.
P values for Grade 3 or more TEAEs, TEAEs leading to discontinuation, and TEAEs leading to death were based on Fisher's exact test.
SAFETY OVERVIEW FOR PATIENTS IN THE RAPID PROGRESSION† SUBGROUPS5
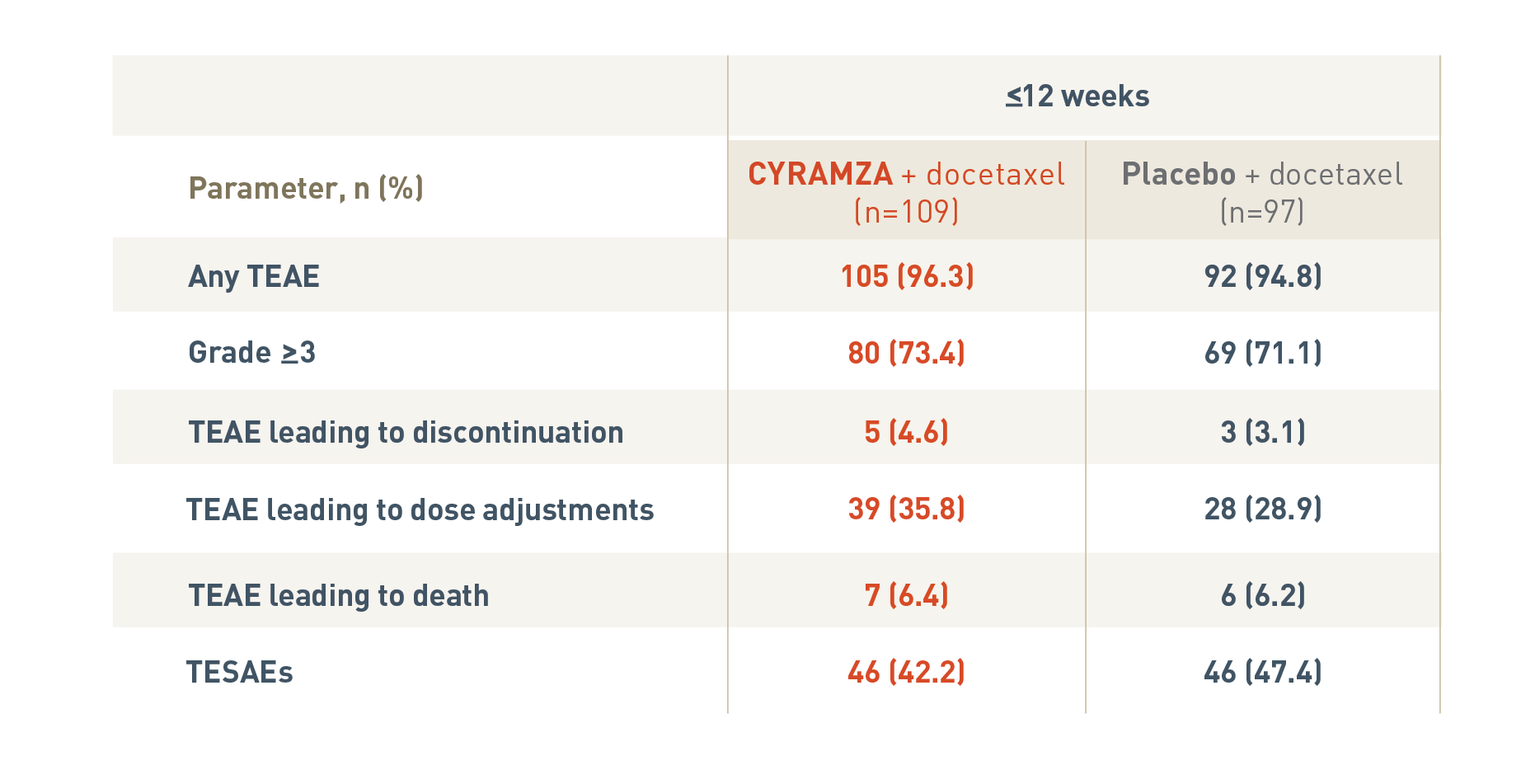
The following safety overview is for patients in the rapid progression subgroup of the REVEL trial at twelve weeks or less.
With CYRAMZA plus docetaxel (n equals 109), 105 patients (96.3 percent) experienced treatment-emergent adverse events (TEAEs), 80 patients (73.4 percent) experienced TEAEs that were Grade 3 or more with a, 5 patients (4.6 percent) experienced TEAEs leading to treatment discontinuation, 39 patients (35.8 percent) experienced TEAEs leading to dose adjustments, 7 patients (6.4 percent) experienced TEAEs leading to death, and 46 patients (42.2 percent) experienced treatment-emergent serious adverse events (TESAEs).
With placebo plus docetaxel (n equals 97), 92 patients (94.8 percent) experienced TEAEs, 69 patients (71.1 percent) experienced TEAEs that were Grade 3 or more, 3 patients (3.1 percent) experienced TEAEs leading to treatment discontinuation, 28 patients (28.9 percent) experienced TEAEs leading to dose adjustments, 6 patients (6.2 percent) experienced TEAEs leading to death, and 46 patients (47.4 percent) experienced TESAEs.
REVEL Exploratory Analyses5,6:
The REVEL trial was not adequately powered or error-controlled for subgroup analyses. No formal hypotheses regarding safety outcomes were tested in the REVEL trial. The analyses described here were post hoc and exploratory.
*Adverse events included TEAEs, Grade ≥3 TEAEs, TESAEs, and TEAEs leading to dose adjustments, discontinuation, or death.5,6
† Rapidly progressing disease is defined as time to progression within 12 weeks after starting initial platinum-based treatment.5
‡ The safety population is defined as patients receiving at least 1 dose of study drug and was based on the actual treatment received.6,8
§ P<0.05; based on Fisher’s exact test.6
Incidence of pulmonary hemorrhage by histology (REVEL trial)1
Pulmonary Hemorrhage by Histology

The following incidence of pulmonary hemorrhage data by histology is from the REVEL trial.
The incidence of all grades pulmonary hemorrhage reported in patients with nonsquamous histology was 7 percent for the CYRAMZA plus docetaxel arm and 6 percent for the placebo plus docetaxel arm.
The incidence of all grades pulmonary hemorrhage reported in patients with squamous histology was 10 percent for the CYRAMZA plus docetaxel arm and 12 percent for the placebo plus docetaxel arm.
The incidence of Grade 3 or more pulmonary hemorrhage reported in patients with nonsquamous histology was 1 percent for the CYRAMZA plus docetaxel arm and 1 percent for the placebo plus docetaxel arm.
The incidence of Grade 3 or more pulmonary hemorrhage reported in patients with squamous histology was 2 percent for the CYRAMZA plus docetaxel arm and 2 percent for the placebo plus docetaxel arm.
Additional adverse events to consider with CYRAMZA as an antiangiogenic therapy7
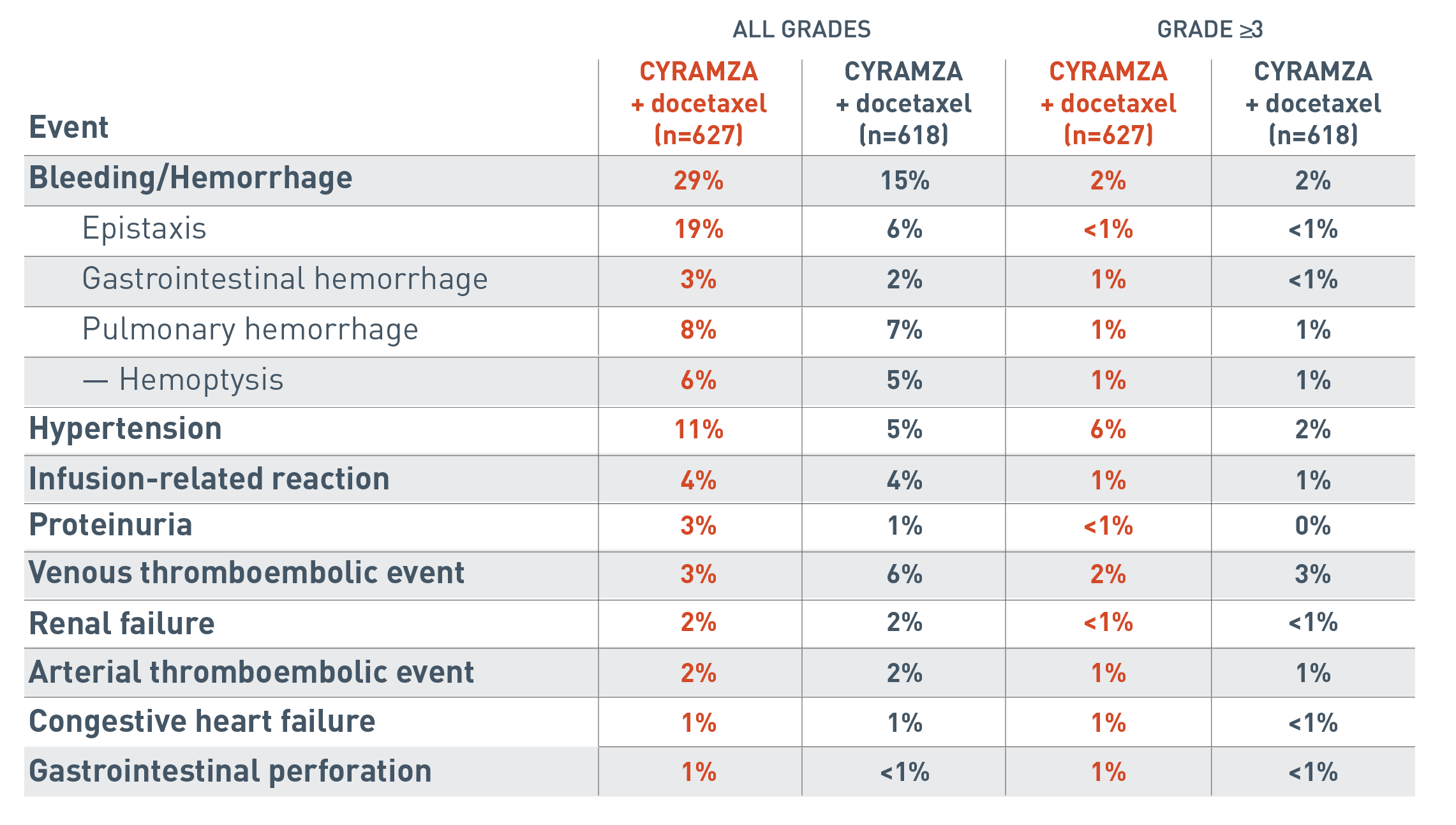
The following additional adverse events associated with antiangiogenic therapy are for the REVEL trial.
The all grades clinically relevant adverse reactions reported for the CYRAMZA plus docetaxel arm (n equals 627) were bleeding/hemorrhage (29 percent), epistaxis (19 percent), gastrointestinal hemorrhage (3 percent), pulmonary hemorrhage (8 percent), hemoptysis (6 percent), hypertension (11 percent), infusion-related reaction (4 percent), proteinuria (3 percent), venous thromboembolic event (3 percent), renal failure (2 percent), arterial thromboembolic event (2 percent), congestive heart failure (1 percent) and gastrointestinal perforation (1 percent).
The all grades clinically relevant adverse reactions reported for the placebo plus docetaxel arm (n equals 618) were bleeding/hemorrhage (15 percent), epistaxis (6 percent), gastrointestinal hemorrhage (2 percent), pulmonary hemorrhage (7 percent), hemoptysis (5 percent), hypertension (5 percent), infusion-related reaction (4 percent), proteinuria (1 percent), venous thromboembolic event (6 percent), renal failure (2 percent), arterial thromboembolic event (2 percent), congestive heart failure (1 percent) and gastrointestinal perforation (less than 1 percent).
The Grade 3 or more clinically relevant adverse reactions reported for the CYRAMZA plus docetaxel arm were bleeding/hemorrhage (2 percent), epistaxis (less than 1 percent), gastrointestinal hemorrhage (1 percent), pulmonary hemorrhage (1 percent), hemoptysis (1 percent), hypertension (6 percent), infusion-related reaction (1 percent), proteinuria (less than 1 percent), venous thromboembolic event (2 percent), renal failure (less than 1 percent), arterial thromboembolic event (1 percent), congestive heart failure (1 percent) and gastrointestinal perforation (1 percent).
The Grade 3 or more clinically relevant adverse reactions reported for the placebo plus docetaxel arm were bleeding/hemorrhage (2 percent), epistaxis (less than 1 percent), gastrointestinal hemorrhage (less than 1 percent), pulmonary hemorrhage (1 percent), hemoptysis (1 percent), hypertension (2 percent), infusion-related reaction (1 percent), proteinuria (0 percent), venous thromboembolic event (3 percent), renal failure (less than 1 percent), arterial thromboembolic event (1 percent), congestive heart failure (less than 1 percent) and gastrointestinal perforation (less than 1 percent).
Ramucirumab inhibited angiogenesis in an in vivo animal model
ITT=intent-to-treat; TEAE=treatment-emergent adverse event; TESAE=treatment-emergent serious adverse event.
References
- CYRAMZA (ramucirumab) package insert. Indianapolis, IN: Eli Lilly and Company; 2021.
- Data on file. Eli Lilly and Company. ONC20150915A.
- Data on file. Eli Lilly and Company. ONC20150915B.
- Supplement to: Garon EB, Ciuleanu T-E, Arrieta O, et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): a multicentre, double-blind, randomised phase 3 trial. Lancet. 2014;384(9944):665-673.
- Reck M, Shepherd F, Pérol M, et al. Effects of second-line ramucirumab after rapid time to progression on first-line therapy: subgroup analysis of REVEL in advanced non-small cell lung cancer. Oral presentation presented at: IASLC 18th World Conference on Lung Cancer; October 15-18, 2017; Yokohama, Japan.
- Data on File, Lilly USA, LLC, DOF-RB-US-0005.
- Garon EB, Ciuleanu T-E, Arrieta O, et al. Ramucirumab plus docetaxel versus placebo plus docetaxel for second-line treatment of stage IV non-small-cell lung cancer after disease progression on platinum-based therapy (REVEL): a multicentre, double-blind, randomised phase 3 trial. Lancet. 2014;384(9944):665-673.
- Data on File, Lilly USA, LLC, DOF-RB-US-0006.